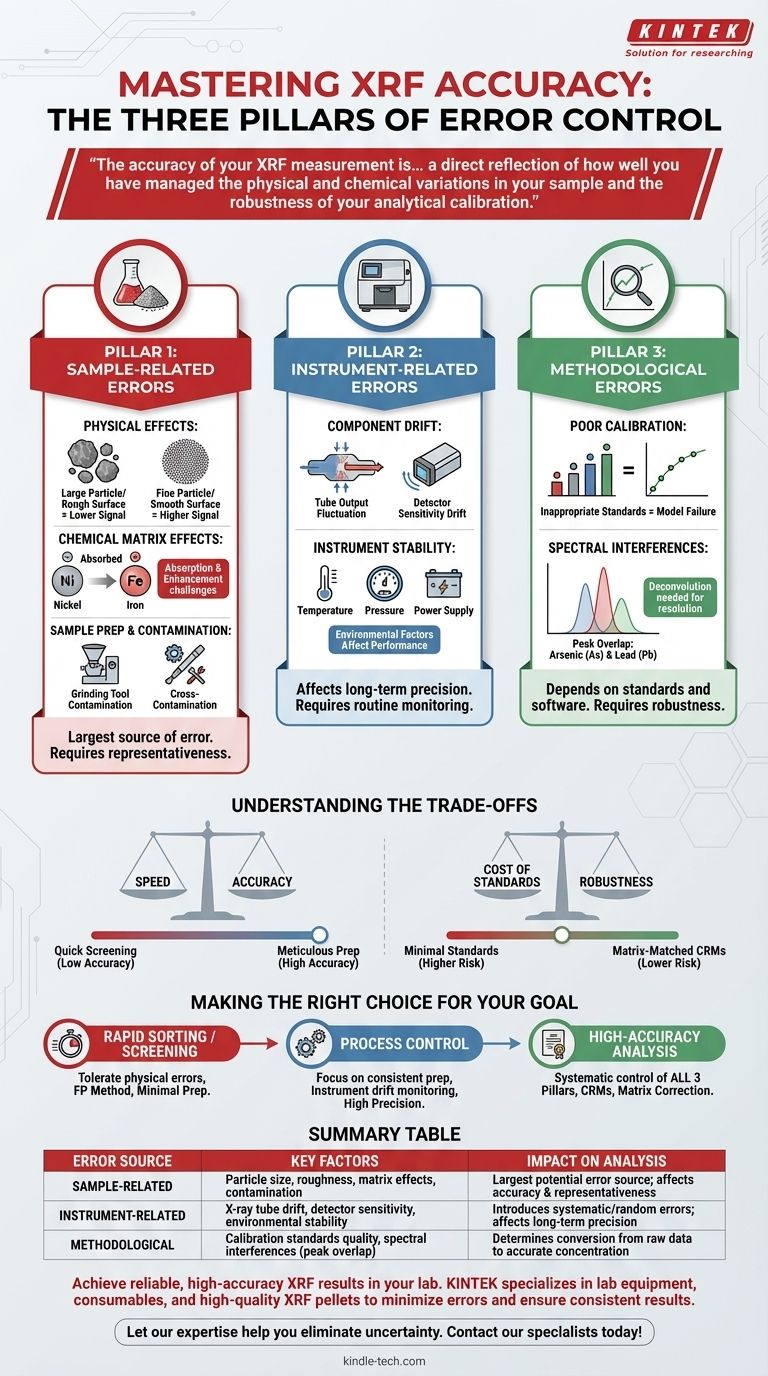
Bei der Röntgenfluoreszenzanalyse (RFA) entstehen Fehler aus drei Hauptbereichen: der Probe selbst (physikalische und chemische Eigenschaften), der Hardware und Stabilität des Instruments sowie der verwendeten Analysemethode oder Kalibrierung. Während Faktoren wie Kontamination der Probe während der Vorbereitung erheblich sind, hängt ein wirklich genaues Ergebnis von der Kontrolle der Variablen in allen drei Bereichen ab.
Die Genauigkeit Ihrer RFA-Messung ist nicht nur eine Funktion der Qualität des Spektrometers, sondern spiegelt direkt wider, wie gut Sie die physikalischen und chemischen Schwankungen Ihrer Probe und die Robustheit Ihrer analytischen Kalibrierung kontrolliert haben.

Die drei Säulen der RFA-Fehler
Um zuverlässige Ergebnisse zu erzielen, müssen Sie verstehen, wo potenzielle Ungenauigkeiten entstehen können. Wir können diese Quellen in drei verschiedene Kategorien einteilen.
Säule 1: Probenbezogene Fehler
Dies ist oft die größte Fehlerquelle. Das Spektrometer kann nur die Probe messen, die ihm vorgelegt wird; wenn die Probe keine wahre Darstellung des Gesamtmaterials ist, werden die Ergebnisse fehlerhaft sein.
Physikalische Effekte
Die physikalische Beschaffenheit der Probe beeinflusst das Röntgenfluoreszenzsignal erheblich. Zu den Schlüsselfaktoren gehören Partikelgröße, Oberflächenrauheit und Probenhomogenität.
Feinere Partikel erzeugen im Allgemeinen ein intensiveres Fluoreszenzsignal als größere. Eine inkonsistente Vermahlung oder eine raue Oberfläche kann zu erheblichen, unvorhersehbaren Fehlern führen.
Chemische Matrixeffekte
Dies bezieht sich darauf, wie andere Elemente in der Probe die Röntgenstrahlen des Elements absorbieren oder verstärken, das Sie messen möchten. Dies ist eine grundlegende Herausforderung in der RFA.
Zum Beispiel absorbiert eine hohe Konzentration an Eisen die Fluoreszenz von Nickel stark, wodurch das Nickel weniger konzentriert erscheint, als es tatsächlich ist. Diese Effekte müssen mathematisch korrigiert werden.
Probenvorbereitung und Kontamination
Die Art und Weise, wie eine Probe vorbereitet wird, ist ein kritischer Kontrollpunkt. Fehler, die hier entstehen, sind irreversibel.
Wie bereits erwähnt, kann Kontamination durch Mahlgeräte Fremdelemente einführen. Ebenso kann es zu Kreuzkontamination zwischen Proben kommen, wenn die Vorbereitungswerkzeuge zwischen den Anwendungen nicht akribisch gereinigt werden.
Säule 2: Instrumentenbezogene Fehler
Obwohl moderne RFA-Spektrometer hochstabil sind, sind sie nicht perfekt. Hardware-Schwankungen können systematische oder zufällige Fehler in die Analyse einführen.
Komponentendrift
Die beiden kritischsten Komponenten, die Röntgenröhre und der Detektor, können im Laufe der Zeit Leistungsänderungen erfahren.
Die Intensität der Röhrenleistung kann schwanken, und die Empfindlichkeit des Detektors kann aufgrund von Temperaturänderungen oder Alterung driften. Diese Änderungen verlaufen in der Regel langsam und können durch routinemäßige Überwachung gesteuert werden.
Instrumentenstabilität
Faktoren wie Umgebungstemperatur, Luftdruck (in einigen Systemen) und Stabilität der Stromversorgung können die Leistung des Spektrometers beeinflussen.
Die Aufrechterhaltung einer kontrollierten Umgebung für das Instrument ist entscheidend für die Erzielung hochpräziser, langfristiger Analyseergebnisse.
Säule 3: Methodische und Kalibrierungsfehler
Selbst bei einer perfekten Probe und einem stabilen Instrument hängt das Endergebnis vollständig von der Analysemethode und der Qualität der Kalibrierung ab.
Schlechte Kalibrierung
Die Kalibrierung ist das mathematische Modell, das rohe Röntgenintensitäten in Elementkonzentrationen umwandelt. Dieses Modell ist nur so gut wie die Standards, die zu seiner Erstellung verwendet wurden.
Die Verwendung unzureichender oder ungeeigneter Kalibrierstandards, die nicht zur chemischen Matrix Ihrer unbekannten Proben passen, ist eine Hauptquelle für erhebliche Analysefehler.
Spektrale Interferenzen
Manchmal liegen die charakteristischen Röntgenlinien von zwei verschiedenen Elementen so nah beieinander, dass der Detektor sie nicht auflösen kann. Dies wird als Peak-Überlappung bezeichnet.
Beispielsweise überlappt die K-alpha-Linie von Arsen mit der K-beta-Linie von Blei. Anspruchsvolle Software ist erforderlich, um diese Peaks mathematisch zu entfalten und ein genaues Ergebnis für jedes Element zu melden.
Die Abwägungen verstehen
Die Kontrolle jeder Fehlerquelle kann zeitaufwändig und kostspielig sein. Der Schlüssel liegt darin, die Sorgfalt bei der Vorbereitung und Analyse an Ihr spezifisches Ziel anzupassen.
Geschwindigkeit vs. Genauigkeit
Eine schnelle „Point-and-Shoot“-Analyse einer unvorbereiteten Probe kann für die einfache Materialidentifizierung oder das Screening ausreichend sein.
Dieser Ansatz opfert jedoch die Genauigkeit und ist für die Qualitätskontrolle oder die Einhaltung gesetzlicher Vorschriften völlig ungeeignet, wo eine akribische Probenvorbereitung (wie das Mahlen und Verpressen von Pellets) nicht verhandelbar ist.
Kosten der Standards vs. Robustheit
Die Erstellung einer robusten Kalibrierung erfordert eine breite Palette hochwertiger, matrixangepasster zertifizierter Referenzmaterialien, was teuer sein kann.
Die Verwendung eines minimalen Satzes von Standards oder die Verlass auf „Typ-Standardisierung“ (Anpassung einer Werkskalibrierung mit ein oder zwei lokalen Proben) ist billiger, birgt aber ein höheres Fehlerrisiko, wenn Ihre Proben von den Standards abweichen.
Die richtige Wahl für Ihr Ziel treffen
Ihre analytische Strategie sollte durch die Frage bestimmt werden, die Sie beantworten müssen.
- Wenn Ihr Hauptaugenmerk auf der schnellen Materialsortierung oder dem Screening liegt: Sie können Fehler aufgrund physikalischer Effekte oft tolerieren und eine einfache Methode der fundamentalen Parameter (FP) verwenden, wodurch die Probenvorbereitung minimiert wird.
- Wenn Ihr Hauptaugenmerk auf der Prozesskontrolle mit einem bekannten Materialtyp liegt: Ihr Hauptanliegen ist die Präzision, daher sollten Sie sich auf eine hochkonsistente Probenvorbereitung und die routinemäßige Überwachung der Instrumentendrift konzentrieren.
- Wenn Ihr Hauptaugenmerk auf hochgenauen Analysen für Zertifizierungen oder Forschung liegt: Sie müssen systematisch alle drei Säulen angehen, indem Sie eine akribische Probenvorbereitung, zertifizierte Referenzmaterialien für die Kalibrierung und Software zur Matrixkorrektur verwenden.
Letztendlich ist die Erzielung von Genauigkeit in der RFA eine Übung in systematischer Kontrolle, bei der das Verständnis der potenziellen Fehlerquellen der erste Schritt zur deren Beseitigung ist.
Zusammenfassungstabelle:
| Fehlerquelle | Schlüsselfaktoren | Auswirkung auf die Analyse |
|---|---|---|
| Probenbezogen | Partikelgröße, Oberflächenrauheit, chemische Matrixeffekte, Kontamination | Größte potenzielle Fehlerquelle; beeinflusst die Genauigkeit und Repräsentativität des Ergebnisses |
| Instrumentenbezogen | Drift der Röntgenröhre, Detektorempfindlichkeit, Umgebungsstabilität | Führt systematische oder zufällige Fehler ein; beeinflusst die langfristige Präzision |
| Methodisch | Qualität der Kalibrierstandards, spektrale Interferenzen (Peak-Überlappung) | Bestimmt die Umwandlung von Rohdaten in genaue Konzentrationswerte |
Erzielen Sie zuverlässige, hochgenaue RFA-Ergebnisse in Ihrem Labor.
Der Weg zu präzisen Analysen erfordert die Kontrolle der Variablen in Bezug auf Ihre Probe, Ihr Instrument und Ihre Methode. KINTEK ist spezialisiert auf Laborgeräte und Verbrauchsmaterialien und bedient Laboranforderungen mit hochwertigen RFA-Pellets, Pressen und Zubehör, die darauf ausgelegt sind, Fehler bei der Probenvorbereitung zu minimieren und konsistente Ergebnisse zu gewährleisten.
Lassen Sie sich von unserer Expertise helfen, Unsicherheiten zu beseitigen. Kontaktieren Sie noch heute unsere Spezialisten, um Ihre spezifische Anwendung zu besprechen und wie wir Ihre analytischen Ziele unterstützen können.
Visuelle Anleitung